Catalog of candidate genes involved in plant-microbe relationships.
As the global population rises, estimated to hit nearly 10 billion by 2050, so does the need to boost crop yields and produce enough plant material for both food and sustainable alternative fuels. To help improve crop breeding strategies and overcome challenges such as making plants more tolerant of marginal lands, and stresses such as drought and low nutrient availability, researchers are focusing on understanding and promoting beneficial plant-microbe relationships.
Published December 18, 2017 issue in Nature Genetics, a team led by researchers at the U.S. Department of Energy (DOE) Joint Genome Institute (JGI), a DOE Office of Science User Facility, and the Howard Hughes Medical Institute at the University of North Carolina at Chapel Hill (UNC) have exploited a catalog of bacterial genomes to identify and characterize candidate genes that aid bacteria in adapting to plant environments, specifically genes involved in bacterial root colonization.
Most of the studies in the field to date have focused on the community structure of the plant microbiome, i.e. “who is there,” and less on the function, i.e. “what they are doing, how and when they are doing it.” Previous studies that have considered function have mainly looked at a single host-microbe interaction, such as the one between an Arabidopsis plant and a pathogen.
“If we want to engineer the right microbiome to support plant growth, we need to understand the real function of the microbiome and not just sequence marker genes,” says study co-first author Asaf Levy, a research scientist at the JGI. “Here we used a massive genomic and computational effort to address the fundamental and important question: ‘How does the plant microbiome interact with the plant?'”
Most of the interaction between microbes and plants occurs at the interface between the roots and soil. Researchers from UNC, Oak Ridge National Lab, and the Max Planck Institute isolated novel bacteria from the root environment of Brassicaceae (191), poplar trees (135), and maize (51). The genomes of these 377 bacterial isolates, plus an additional 107 single bacterial cells from roots of A. thaliana, were then sequenced, assembled, and annotated at the JGI.
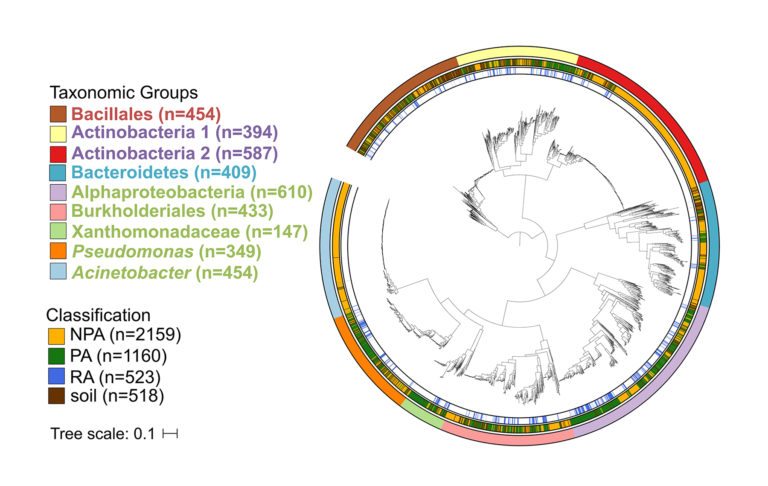
The authors then combined the new genomes with thousands of publicly available genomes that represent the major groups of plant-associated bacteria, and included bacteria from multiple plant and non-plant environments, such as the human gut, for comparison. The resulting database of 3837 genomes, 1160 of which are from plants, was used in a comparative genomics analysis.
The researchers then identified genes that are enriched in the genomes of plant-associated and root-associated organisms.
“It’s very important for us to understand what genes and functions microbes use to colonize plants because only then might we have a chance to rationally devise useful ‘plant probiotics’ to help us raise more food and energy crops with fewer chemical inputs such as fertilizers and pesticides or fungicides,” said study senior author Jeff Dangl, a Howard Hughes Medical Institute investigator and the John N. Couch Professor of Biology at the University of North Carolina at Chapel Hill
Among the key insights gained from the study was that plant- and soil-associated genomes tend to be larger than control genomes from the same clade. This was found to be due in part to enrichment of genes involved in sugar metabolism and transport, likely an adaptation to photosynthesis-derived plant carbon, generated by nature’s “candy factories,” says Levy. Up to 20 percent of the carbon fixed by plants through photosynthesis is exuded through the roots as sugars to attract microbes.
Numerous genes that seem to mimic plant functions—by encoding “Plant-Resembling PA and RA Domains” or PREPARADOs—were also identified. “It is well known that plant pathogens use proteins that mimic plant domains required for immune function,” said Dangl. “Imagine that the pathogen injects directly into the plant cell a protein that mimics part of a particular immune system machine. It’s like putting a partly defective cog into a wheel—the wheels can’t turn anymore. We reckon that the plant-associated protein domains that we identified might work in the same way.”
Rapidly evolving genes are often a signature of a molecular arms race between organisms sharing an environment. These genes are often used in offense or defense against another organisms. Two new rapidly evolving protein families associated with different “lifestyles” of related plant-associated bacteria were identified in the study. One, found in commensal bacteria, was dubbed “Jekyll”; the other, found in pathogenic bacteria, was named “Hyde.” With collaborators from Virginia Tech and ETH (Switzerland), JGI scientists discovered that the latter are very efficient in killing competing bacteria, potentially to help these “Hydes” take over the leaf niche. Berkeley Lab’s Innovation and Partnerships Office (IPO) has filed a patent application for this family as a potential antibacterial mechanism for controlling phytopathogens.
The complete catalog of new genomes and plant-associated genes is available to the research community through a dedicated web portal: Genomic Features of Bacterial Adaptation to Plants.
“The database is a precious resource for the research community studying plant-microbe interactions as it is an unbiased way to identify potentially interesting genes involved in interaction with a plant—including many totally novel genes. We are currently experimentally studying the function of many of these genes to gain a better functional understanding of the plant microbiome.” Levy says.
Collaborating institutions on this work included Oak Ridge National Laboratory, University of Washington, and the Max Planck Institute. In addition to the DOE Office of Science, funding for this work was provided through the NSF INSPIRE Program to Jeff Dangl (UNC), Ruth Ley (MPI) and Susannah Tringe (JGI). Additional funding was provided by the DOE-USDA Feedstocks Program to the Dangl lab and Dale Pelletier (ORNL).
Source: Joint Genome Institute