Soybean is one of the most important crops worldwide. A high-quality reference genome will facilitate its functional analysis and molecular breeding. Previously, biologists from China (Chinese Academy of Science, University of Science and Technology of China, Jiangsu Academy of Agricultural Sciences, Berry Genomics Corporation) de novo assembled a high-quality Chinese soybean genome Gmax_ZH13 (Shen et al., 2018). However, due to technical limitations, a large number of small contigs were not anchored onto chromosomes.
Recently, the leader research group for Gmax_ZH13 genome project from the Institute of Genetics and Developmental Biology, Chinese Academy of Science, updated the Gmax_ZH13 genome to a golden reference genome Gmax_ZH13_v2.0.
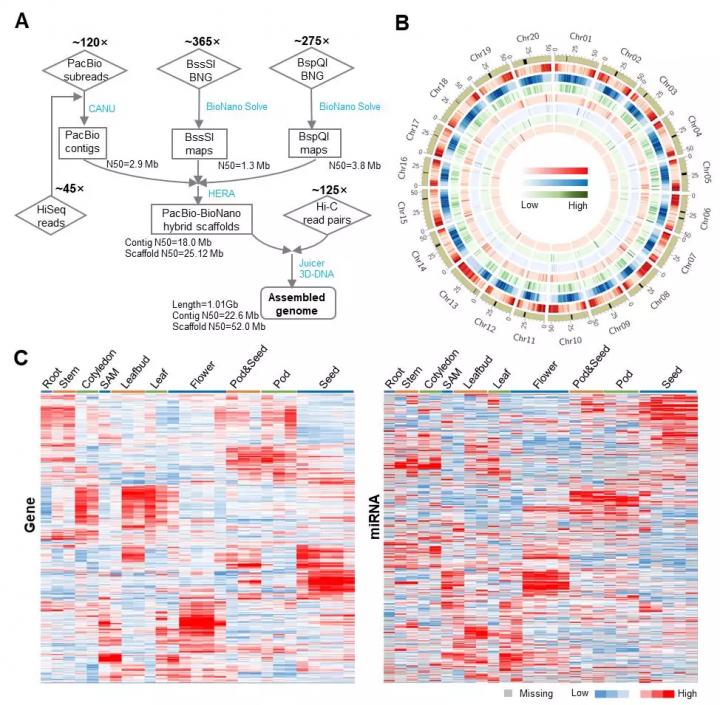
Based on the Gmax_ZH13, by adding more sequence data and refresh assembly pipeline (Figure 1A), researchers finally assembled Gmax_ZH13_v2.0 with a length of 1,011,174,350 bp. Its assembly quality was increased dramatically. When compared to Gmax_ZH13, the Contig N50 size of Gmax_ZH13_v2.0 increased 6.5 times (from 3.46 Mb to 22.6 Mb), gap number decreased 1.8 times (from 815 to 448) and gap length decreased 8.8 times (from 20.49 Mb to 2.33Mb). Meanwhile, the un-anchored contig number decreased 17 times (from 549 to 36), resulting in the ratio of sequence that anchored to 20 chromosomes reaching 98%. All these assembly parameters indicated the high completeness of Gmax_ZH13_v2.0. Besides nuclear chromosomes, researchers assembled the circular genomes of chloroplast and mitochondria with a length of 152,220 bp and 513,779 bp respectively.
To improve the accuracy of gene annotation, in addition to Iso-seq reads used for Gmax_ZH13 annotation, researches performed RNA-seq and smRNA-seq for another 27 ZH13 samples, which were collected from different tissues at different developmental stages. They finally annotated 55,443 protein coding genes containing 96,366 mRNAs in the nuclear genome, 81 protein coding genes in the chloroplast genome and 49 protein coding genes in the mitochondrial genome. 97% of the 1,440 single copy Embryophyta genes in BUSCO_v3 were completely assembled, confirming the high quality of protein coding gene annotation. Besides that, non-coding genes were also annotated, including 297 rRNA, 1,112 tRNA, 166 snRNA 1,816 snoRNA and 35926 TE. Especially, 331 MIRNA genes and the mature miRNAs they produced were annotated by smRNA-seq data (Figure 1B).
Researchers also provided a detailed expression profiling for all protein coding genes and miRNAs they annotated (Figure 1C). These expression profiling data will be helpful for soybean fundamental research, for instance, searching expression pattern of individual genes or choosing tissue specific expression genes. Moreover, the data can be used to investigate the relationship of miRNAs and their target genes because they came from the same sample sets.
“We updated the Gmax_ZH13 genome to a more complete and continuous platinum reference genome Gmax_ZH13_ v2.0, did comprehensive annotation and provided detailed expression information for it”, said Professor Zhixi Tian, the leader of the Gmax_ZH13 Chinese soybean genome project. “We believe that the new genome will greatly facilitate soybean fundamental research and molecular breeding.”
Source: Science China Press